
When you pick up a prescription today, there’s a better than 90% chance it’s a generic drug. That’s not just convenience-it’s the result of a 200-year battle over safety, cost, and control in American medicine. Most people don’t realize that the pills they take for high blood pressure, diabetes, or antibiotics were once nearly impossible to find outside brand-name versions. The story of how generics became the backbone of U.S. healthcare isn’t about science alone-it’s about laws, lobbying, scandals, and a quiet revolution in how we pay for medicine.
The Roots of Drug Standards
Long before modern pharmacies, medicine was a gamble. In the early 1800s, there was no way to tell if a pill contained the right ingredient or was just flour and dye. In 1820, eleven doctors met in Washington, D.C., and created the first U.S. Pharmacopeia-a list of approved drugs and their formulas. This wasn’t a law, but it was the first attempt to standardize what counted as real medicine. By 1888, the American Pharmaceutical Association added the National Formulary, which helped pharmacists avoid counterfeit drugs. Still, anyone could sell anything labeled as “quinine” or “morphine,” and many didn’t.The First Real Protections
The turning point came in 1906 when President Theodore Roosevelt signed the Federal Food and Drugs Act. It didn’t ban dangerous drugs-it just forced companies to list ingredients on the label. That sounds small, but it was the first time the government said: “If you’re selling medicine, you have to tell people what’s in it.” The law was sparked by public outrage over adulterated products, but it wasn’t enough. In 1937, over 100 people died after taking a liquid form of sulfanilamide that used diethylene glycol-a toxic antifreeze ingredient. The public outcry led to the 1938 Federal Food, Drug, and Cosmetic Act, which required drugmakers to prove their products were safe before selling them. For the first time, the FDA had real power to stop dangerous drugs from reaching shelves.Proving That Drugs Actually Work
The next big shift came in 1962, after the thalidomide tragedy overseas. Although the drug wasn’t approved in the U.S., its effects in Europe showed how deadly untested drugs could be. The Kefauver-Harris Drug Amendments forced every drug on the market since 1938 to prove it wasn’t just safe-but effective. That meant thousands of drugs, including many older ones, had to go back to the lab. It also created the framework for what we now call the “new drug application.” But here’s the catch: the law didn’t say anything about generics. Brand-name companies still held all the data. If you wanted to make a copy, you had to start from scratch with expensive clinical trials. That made generics rare and expensive.
The Hatch-Waxman Act Changed Everything
In 1984, everything changed. The Drug Price Competition and Patent Term Restoration Act, better known as the Hatch-Waxman Act, created the modern system for generic drugs. Before this, only about 19% of prescriptions were for generics. After? That number jumped fast. The law did two big things. First, it created the Abbreviated New Drug Application (ANDA). Instead of repeating full clinical trials, generic manufacturers only had to prove their version was bioequivalent-meaning it delivered the same amount of active ingredient at the same rate as the brand-name drug. Second, it gave brand-name companies a 5-year exclusivity period after approval, plus up to 5 more years if they had patents on the drug’s use. This wasn’t a giveaway-it was a trade. Generics got a faster path to market. Innovators got extra time to recoup their R&D costs.The Rise of the Generic Market
The results were dramatic. By 2022, generics made up 90.5% of all prescriptions filled in the U.S. But they only cost 23.4% of total drug spending. That’s because they’re cheaper-often 80-85% less than the brand-name version. In 2021 alone, generic drugs saved the U.S. healthcare system $373 billion. Over the last decade, that adds up to more than $3.7 trillion in savings. The Centers for Medicare & Medicaid Services, the VA, and private insurers all rely on generics to keep costs down. Without them, millions of people couldn’t afford their medications.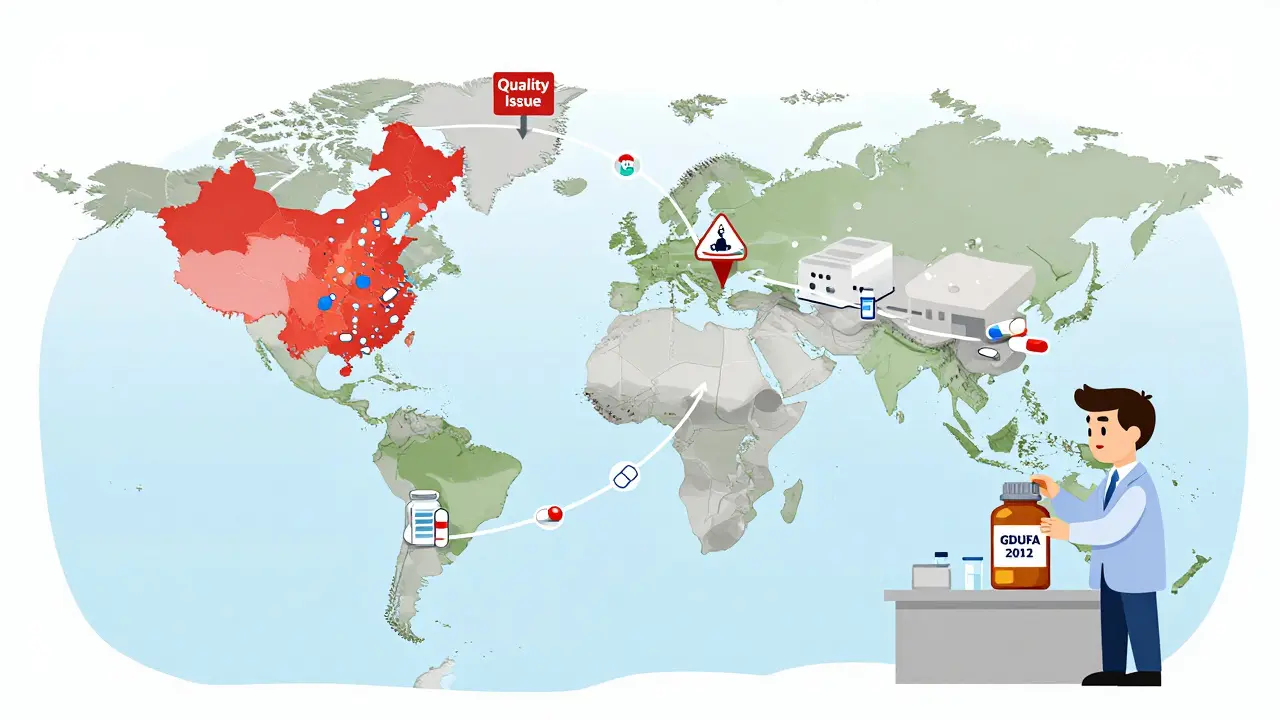
Where Things Got Messy
But the system isn’t perfect. One loophole in Hatch-Waxman lets brand-name companies sue generic makers, triggering a 30-month delay in approval. This tactic, called “evergreening,” has been used hundreds of times to block competition. In 2019, Congress passed the CREATES Act to stop this. It bans brand-name companies from refusing to sell samples to generic manufacturers, which they used to delay testing. The FDA has already taken 27 enforcement actions under this law. Another problem? Quality. Between 2018 and 2022, 65% of the 1,234 drug shortages in the U.S. involved generic drugs. Why? Because most of the active ingredients come from factories in China and India. About 80% of API facilities are overseas. When one factory has a problem-like contamination or inspection failure-it can ripple across dozens of medications. The FDA has been trying to fix this since 2007 with the Generic Initiative for Value and Efficiency (GIVE), and later with GDUFA in 2012. Since then, review times for generic applications dropped from 30 months to 10 months, and approval rates jumped from 45% to 95%.The Price Volatility Problem
Here’s something counterintuitive: even though generics are cheaper overall, prices for some can spike overnight. Between 2013 and 2017, 15% of generic drugs saw price increases over 100%. Why? Because when a drug has only one or two manufacturers, competition disappears. A single plant shutdown or regulatory issue can turn a $1 pill into a $50 pill. The FDA has flagged dozens of these cases, especially for older drugs like doxycycline, tetracycline, and insulin. In some cases, prices rose because manufacturers stopped making them-then restarted production when demand spiked.What’s Next?
The next frontier isn’t just pills-it’s biosimilars. These are generic versions of complex biologic drugs, like those used for cancer or autoimmune diseases. They’re harder to copy than simple chemical pills, but the same logic applies: if we can make them affordable, we can save billions. Companies are already working on biosimilars for Humira, Enbrel, and other high-cost drugs. The FDA approved its first biosimilar in 2015. By 2027, analysts expect biosimilars to make up a growing share of the market.Today, the FDA oversees more than 22,000 generic drug products and 13,000 manufacturing sites worldwide. Roughly 900 ANDAs are approved every year. The system isn’t flawless, but it works. Without the history of laws, scandals, and reforms, most Americans wouldn’t be able to afford their prescriptions. The next challenge? Making sure the system stays fair, transparent, and resilient-not just for today, but for the next 100 years.
What is an ANDA?
An ANDA stands for Abbreviated New Drug Application. It’s the paperwork a generic drug company submits to the FDA to get approval to sell a copy of a brand-name drug. Instead of repeating full clinical trials, the company must prove its version is bioequivalent-meaning it works the same way in the body as the original. This is the key reason generic drugs are cheaper and faster to bring to market.
Why are generic drugs so much cheaper?
Generic drugs are cheaper because manufacturers don’t have to pay for expensive clinical trials. The original brand-name company already proved the drug is safe and effective. Generic makers only need to show their version delivers the same amount of active ingredient at the same rate. This cuts development costs by 80-90%. They also face more competition, which keeps prices low.
Are generic drugs as safe and effective as brand-name drugs?
Yes. The FDA requires generic drugs to have the same active ingredient, strength, dosage form, and route of administration as the brand-name version. They must also meet the same strict standards for purity, potency, and quality. Studies show generics perform the same in the body. The only differences are in inactive ingredients like fillers or dyes, which don’t affect how the drug works.
Why do some generic drugs have shortages?
Many generic drug ingredients come from factories overseas, mostly in China and India. When one plant has a quality issue, gets shut down by regulators, or faces supply chain delays, it can affect dozens of medications. Also, if a drug has only one or two manufacturers, and one stops production, supply can collapse. The FDA tracks these shortages and works to find alternative suppliers, but it’s not always fast enough.
Can brand-name companies block generic competition?
Yes, but only under certain rules. Before the CREATES Act, brand-name companies sometimes refused to sell samples to generic makers or restricted distribution to delay testing. They could also file lawsuits to trigger a 30-month delay in FDA approval. These tactics were common and legal under Hatch-Waxman. The CREATES Act now makes these practices illegal, and the FDA has taken enforcement actions against companies that violate them.